

1. INTRODUCTION
Le Manuel Qualité (MAQ) présente les dispositions générales adoptées et mises en œuvre par le laboratoire AL MANAR pour obtenir et garantir la qualité de ses prestations conformément aux exigences de la réglementation, aux exigences de la norme ISO 15189.
Il décrit notamment l’organisation du laboratoire, sa cartographie des processus, ses différents types de prestations et les dispositions mises en place et appliquées systématiquement en matière d’assurance de la qualité.
Il s’adresse à notre structure interne et aux clients du laboratoire (patients, prescripteurs, correspondants, partenaires des établissements de santé et des auditeurs).
Ce document est connu et appliqué par chaque personne travaillant au laboratoire.
2. DOMAINE D’APPLICATION
Le système de management de la qualité est organisé selon les exigences réglementaires de la norme ISO 15189:2012.
Ce système couvre l’ensemble des activités du laboratoire de biologie médicale. Les processus du laboratoire sont classés en trois grandes familles :
-
Les processus de réalisation : processus pré analytique, analytique et post analytique
-
Les processus de pilotage – Management du SMQ
-
Les processus supports – Management des ressources
3. RÉFÉRENCES – DOCUMENTATION
Principaux textes réglementaires applicables à l’activité
Contrats de coopération
Normes: Norme NM EN ISO 15189:2012 Laboratoire de biologie médicale – Exigences particulières concernant la qualité et la compétence
4. DÉFINITIONS ET ABRÉVIATIONS
-
-
Définitions
-
Analyses: Examens biologiques qui concourent au diagnostic, au traitement ou à la prévention des maladies humaines ou qui font apparaître toute autre modification de l’état physiologique
Comptes rendus de résultats : Documents écrits, validés et signés par le biologiste comportant les résultats d’analyses qualitatifs et/ou quantitatifs accompagnés de commentaires aussi souvent qu’il est nécessaire.
Contrôle interne de qualité (CIQ) Ensemble des opérations et activités opérationnelles d’un site de production mises en œuvre pour assurer que les exigences relatives à la qualité des services sont satisfaites.
Evaluation externe de la qualité ou Contrôle Externe de Qualité (CEQ – EEQ) : Vérification des résultats de mesures ou d’observations effectuées en un lieu donné, par comparaison avec les résultats obtenus par d’autres sites sur le même échantillon émis par une entité extérieure qui procède également à l’analyse statistique des données.
Echantillon biologique : Echantillon obtenu par l’acte de prélèvement et sur lequel vont être effectuées une ou plusieurs analyses de biologie médicale.
Qualification du personnel : Validation d’une ou plusieurs compétences techniques acquises.
Valeurs de référence: Résultats obtenus pour un constituant donné dans une population de référence dont les individus sont exempts de pathologie ou de traitement susceptibles de modifier ceux-ci.
Validation des résultats d’analyse : Opération permettant d’assurer qu’un résultat a été obtenu dans des conditions techniques satisfaisantes et est compatible avec le dossier biologique du patient. Cette validation est à la fois technique et biologique.
La validation technique : comporte la vérification de la conformité des conditions d’exécution aux procédures et tient compte notamment des résultats obtenus sur les échantillons de contrôle.
La validation biologique : est le contrôle de la vraisemblance et de la cohérence de l’ensemble des résultats des analyses effectuées pour une personne compte tenu de son état clinique, des traitements thérapeutiques suivis et des résultats antérieurs.
-
-
Abréviations :
-
5. SYSTÈME DE MANAGEMENT DE LA QUALITÉ
-
-
Exigences générales (Chap. 4.1 – 4.2 – ISO 15189)
-
Le laboratoire AL MANAR adopte les exigences de la norme 15189 :2012 pour la mise en œuvre du système de management de la qualité et son amélioration continue.
Cf. cartographie des processus
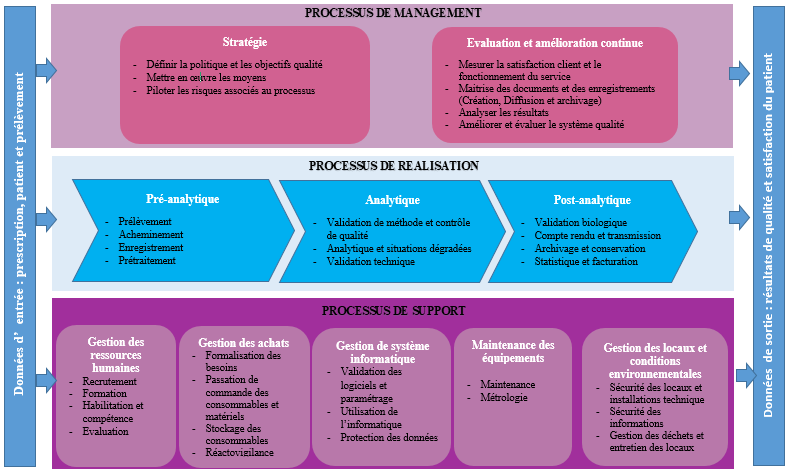
Les processus nécessaires pour le système de management de la qualité sont décrits et comportent pour chaque processus : La finalité, le (les) pilote(s), la description (étapes), les principaux documents associés, l’analyse de risque et les actions préventives, les modalités de surveillance et d’évaluation (indicateurs qualité). Les objectifs et indicateurs qualité suivent la même logique par processus.
|
FP M O1 |
Stratégie |
|
FP M 02 |
Évaluation et amélioration continue |
|
FP R 01 |
Pré-analytique |
|
FP R 02 |
Analytique |
|
FP R 03 |
Post-analytique |
|
FP S 01 |
Gestion des ressources humaines |
|
FP S 02 |
Gestion des achats |
|
FP S 03 |
Gestion de système informatique |
|
FP S 04 |
Maintenance des équipements |
|
FP S 05 |
Gestion des locaux et conditions environnementales |
|
PROCESSUS |
Norme ISO 15189 :2012 |
DOCUMENTATION Liste des procédures (1) |
|
|
Processus management |
|||
| Stratégie |
4.1 – 4.2 |
||
| Evaluation et amélioration continue |
4.3 – 4.8 – 4.9 – 4.10 – 4.11 – 4.12 -4.13 – 4.14 – 4.15 |
||
|
Processus de réalisation – processus métier |
|||
| Pré-analytique | Demandes d’examens
– accueil -revue de contrat |
5.4 |
|
|
Prélèvements Traitement échantillons |
|||
|
Analytique |
Réalisation des analyses Validation technique Validation analytique |
5.5 5.6 |
|
|
Post analytique |
Validation biologique Conservation – élimination échantillons |
5.7 |
|
|
Transmission des comptes rendus de résultats |
5.8 – 5.9 |
||
|
Processus supports ou de soutien |
|||
|
Gestion des ressources humaines |
4.1 – 5.1 |
||
|
Gestion des achats |
4.5 4.6 |
||
|
Gestion de système informatique |
5.10 |
||
|
Maintenance des équipements |
5.3 |
||
|
Gestion des locaux et conditions environnementales |
5.2 – 5.1 |
||
Les procédures et documents relatifs au système de management de la qualité sont identifiés de manière univoque conformément aux exigences de la norme ISO 15189 – 4.3 Maîtrise des documents.
-
-
Documentation (Chap. 4.2 – 4.3 – ISO 15189)
-
Le manuel qualité (Chap. 4.2 – ISO 15189)
-
-
Le responsable qualité du laboratoire veille à la bonne gestion du manuel qualité. Il est responsable de sa rédaction ainsi que de sa révision et mise à jour. La vérification et approbation du Manuel Qualité sont sous la responsabilité de la direction du laboratoire.
La diffusion et l’archivage sont sous la responsabilité de responsable qualité. Le manuel qualité est découpé en chapitres, il comporte un numéro de version et une date d’application, une pagination x/y.
Le manuel qualité est tenu à jour par révision périodique sous l’autorité et la responsabilité du responsable qualité, selon les dispositions de la procédure de maîtrise des documents et des enregistrement.
L’ensemble du personnel est destinataire du manuel qualité, des documents référencés qui les concernent et des exigences relatives à leur mise en œuvre.
-
-
-
Maîtrise des documents (Chap. 4.3 – ISO 15189)
-
-
Structure du système documentaire : La maîtrise du système qualité et tous les processus du laboratoire reposent sur la mise en place de documents qualité classés par niveau selon une pyramide documentaire :
Tous les documents qualité du laboratoire sont répertoriés. Le laboratoire utilise un logiciel qualité avec lequel la gestion documentaire est progressivement organisée de façon maîtrisée (transfert des documents qualité).
La documentation externe est gérée et comporte :
- les documents administratifs (ex : autorisations, …)
- les documents réglementaires applicables au laboratoire.
- les documents techniques : catalogues fournisseurs, manuels techniques, notices techniques des réactifs et des analyses de laboratoire.
- la ou les normes, les documents contractuels et guides
La documentation externe applicable au laboratoire et validée par le directeur du laboratoire ou le responsable qualité (selon la nature des documents). Le suivi et la mise à jour de celles-ci sont assurés par les techniciennes référentes du laboratoire. Se reporter aux dispositions de la procédure élaboration et gestion des documents.
Les informations d’origine externe constituent une ressource fondamentale pour le développement continu des connaissances.
6. RESPONSABILITE DE LA DIRECTION
-
- Engagement de la direction (Chap. 4.1.2 – ISO 15189)
La direction du laboratoire AL MANAR s’engage à :
-
- Ecoute client (Chap. 4.7 – 4.8 – ISO 15189)
L’écoute des clients du laboratoire AL MANAR est assurée par le recueil régulier d’un faisceau d’informations :
- Eléments de satisfaction ou réclamation
- Prestations de conseils
- Exigences spécifiées : délais, degré d’urgence, destinataires, renseignements cliniques, …
Des enquêtes de satisfaction sont effectuées régulièrement auprès des différentes catégories de clients (patients, médecins…), analysées et exploitées pour dégager des axes d’améliorations. Les résultats des enquêtes sont présentés en revue de direction et communiqués en interne.
Les méthodes utilisées pour être à l’écoute des clients (questionnaire, entretien …) tiennent compte des différents types de clients, des objectifs fixés, dans le respect des règles déontologiques de la profession.
-
- Politique qualité (Chap. 4.2 – ISO 15189)
- Planification (Chap. 4.2 – ISO 15189)
- Objectifs qualité
Les objectifs qualité sont déterminés en cohérence avec l’énoncé de la politique qualité. Ils sont formalisés, analysés et ajustés au cours des réunions de qualité. Ils sont déclinés au niveau des activités, des processus et des fonctions concernées pour leur mise en œuvre. L’atteinte des objectifs qualité est vérifiée au cours des réunions de revue de direction à partir des résultats des indicateurs de qualité et corrigé dans un PV de réunion.
-
-
- Planification du système de management de la qualité (Chapitre 4.10-4.11-4.13 ISO 15189)
-
Pour atteindre de manière efficiente les objectifs qualité, le laboratoire planifie des actions d’amélioration aux processus de réalisation des analyses, aux processus de soutien nécessaires à cette réalisation et aux processus de management.
La direction du laboratoire veille au maintien de la cohérence du système de management de la qualité.
Nos efforts sur la validation des méthodes d’analyses sont à entretenir pour les portées accréditées.
Cette planification s’organise à partir des éléments suivants :
Planification de la Qualite
-
Renforcement du savoir-faire et des connaissances
-
Ajustement des ressources (aménagement des locaux, personnel, matériels et fournitures)
-
Amélioration des méthodes et de l’organisation
-
Suivi de nouveaux indicateurs de qualité
-
Objectifs qualité du laboratoire
-
Besoins et attentes des clients
-
Exigences réglementaires normes
-
Performance des processus
-
Adéquation du produit et des services associés
-
Non conformités et incidents
-
Adéquation de la gestion documentaire
-
Evaluation des fournisseurs
Le Responsable Qualité est chargé de vérifier et de suivre le bon déroulement et l’organisation de la planification du système de management de la qualité.
Les résultats de la planification de la qualité sont analysés au cours des revues de direction. Des ajustements éventuels sont décidés par la direction sous forme de plans d’amélioration.
-
- Responsabilité de la direction et communication (Chap. 4.1 – ISO 15189)
La direction du laboratoire ALMANAR est chargée de définir et de communiquer la politique et les objectifs qualité à l’ensemble de l’équipe du laboratoire.
Pour déployer la politique et les objectifs qualité la direction du laboratoire a défini une organisation du système qualité faisant intervenir :
- Le biologiste directeur
- L’Assistant biologiste – Le responsable qualité
- Les référents par secteur d’activité :
- référents secteur technique (sous forme de qualification – habilitation) :
- Technicien référent hématologie
- Technicien référent hémostase
- Technicien référent biochimie
- Technicien référent bactériologie
- Technicien référent mycologie
- référent métrologie
- référent secrétariat accueil
- référent prélèvements – infirmières
- référent hygiène et sécurité
Les responsabilités et autorités sont décrites dans les fiches de fonction.
Les qualifications et habilitations du personnel sont organisées, documentées et donnent lieu à des enregistrements permettant de prouver et suivre les aptitudes, les responsabilités.
Processus – 10 – Gestion des ressources humaines
Procédures associées :
ORGANIGRAMME FONCTIONNEL ET HIERARCHIQUE
(Chap. 5.1 – ISO 15189)
Organisation de la communication interne :
Elle repose sur des réunions régulières, en interaction, qui impliquent toute l’équipe du laboratoire. Cette communication interne entretient la dynamique de management de la qualité en faisant intervenir toutes les fonctions du laboratoire.
Remontées » Remontées « Terrain » Non conformités Réclamations Suggestions (enquêtes, personnel)
Comptes rendus « secteurs »
Plan d’actions
Diffusion secteur
- Accueil – secrétariat
- Prélèvements
- Techniques
Processus – 01 Stratégie
-
- Revue de direction (Chap. 4.15 – ISO 15189)
L’organisation de revues de direction permet de mesurer l’efficacité du système de management de la qualité mis en œuvre (ISO 15189:2012 chap. 4.15). La périodicité est annuelle.
Ces réunions sont organisées par la direction du laboratoire, le responsable qualité à partir des données desdifférents éléments d’entrée de la revue (non conformités produits et processus, réclamations, rapports et résultats des audits internes, état d’avancement des actions correctives et préventives, enquêtes de satisfaction et retour d’informations clients, le compte rendu de la revue de direction précédente, les modifications de la réglementation à prendre en compte, les propositions d’amélioration). L’équipe du laboratoire participe, par secteur, à la préparation et la présentation des données de revue de direction.
La direction entérine les actions d’amélioration à mettre en œuvre en cohérence avec la politique et les objectifs qualité du laboratoire.
L’ordre du jour de la revue de direction reprend les exigences de la norme ISO 15189 :2012.
Le compte rendu de réunion validé par la direction du laboratoire comporte les conclusions et décisions communiquées à l’ensemble du personnel du laboratoire.
Les décisions sont formalisées dans un plan d’amélioration.
Processus – 02 – AMELIORATION CONTINUE
Procédures associées :
7. MANAGEMENT DES RESSOURCES
Les exigences de formation, compétences pour diriger un laboratoire relèvent du domaine réglementaire ainsi l’organisation des locaux et les exigences d’hygiène et de sécurité.
-
-
Ressources humaines – Le personnel (Chap. 5.1 – ISO 15189)
-
La gestion du personnel est centrée sur la responsabilisation, le développement des compétences, l’implication de chaque acteur dans le management de la qualité et la prévention des risques. Cette gestion se traduit par une forte communication en interne : réunions de secteur, réunions qualité, participation active aux revues de direction.
Le recrutement de personnel s’appuie sur les définitions de fonction, les qualifications (niveau de compétence requis) en vue de répondre aux besoins immédiats et futurs. Le recrutement est conduit par le directeur du laboratoire en collaboration avec l’équipe du secteur concerné.
Un dossier individuel du personnel est ouvert et tenu régulièrement à jour.
Toute embauche définitive est précédée d’une période d’essai au cours de laquelle le directeur du laboratoire veillera aux points suivants :
-
Les connaissances générales
-
La pratique professionnelle
-
L’aptitude technique ainsi que la capacité d’adaptabilité à différents types de matériels
-
L’aptitude à s’intégrer à l’équipe
-
Le respect des procédures et des règles en vigueur et du code d’éthique de la profession.
-
La compréhension et l’adaptation à la Politique Qualité mise en place au sein de l’entreprise
A la fin de la période d’essai, la direction décide alors de l’intégration de la personne dans le laboratoire.
Chaque personne du laboratoire se voit attribuer une ou plusieurs fonctions décrivant les missions principales, attributions spécifiques à la fonction occupée.
Une évaluation est effectué par le directeur du laboratoire avec chaque salarié pour formaliser des objectifs et identifier les besoins de formation.
La formation continue du personnel est définie dans un plan annuel de formation validé par le biologiste directeur en collaboration avec le biologisteassistant à partir :
-
des projets prioritaires du laboratoire,
-
des besoins exprimés par le personnel,
-
des besoins de maintien à niveau des connaissances et des compétences du personnel.
Des formations hors plan peuvent être réalisées si nécessaire. Les formations sont réalisées par des organismes externes ou par les personnes habilitées en interne. Le suivi de la réalisation du plan est sous la responsabilité du biologiste assistant. Les attestations de formation externe ou interne sont suivies et classées dans les dossiers du personnel par l’assistante qualité.
Les qualification et habilitations sont documentées et validées par les biologistes pour les tâches et les activités qui nécessitent des compétences ou des autorisations particulières et par le responsable qualité (métrologie, audit interne).
Tout salarié du laboratoire est soumis au respect de la confidentialité et du secret professionnel. Un engagement de confidentialité est signé par chaque personne et est archivé dans le dossier du personnel.
Processus — RESSOURCES HUMAINES
Procédures associées :
-
-
Locaux environnement – Matériel (Chap. 5.2 – 5.3 – 5.7 – ISO 15189)
-
Le laboratoire satisfait aux exigences réglementaires en matière de superficie allouée aux activités techniques, au secteur d’activité nécessitant un confinement particulier (prévention des risques de contamination croisée), aux contrôles des accès et au stockage des DASRI avant leur élimination (local dédié). Les sols, surfaces de travail et sanitaires sont désinfectés selon des pratiques conformes à la profession. Des contrôles bactériologiques des surfaces sont organisés régulièrement.
Cette démarche relève de l’analyse de risque (notamment risque de contamination croisée et risque microbiologique plus largement) avec mise en place d’une démarche préventive sous forme de contrôles de surfaces réguliers, planifiés.
L’accès au laboratoire et la circulation à l’intérieur sont limités et définis par affichage. Tout visiteur ou intervenant extérieur s’identifie dans un registre (date, nom, signature). Toutes les installations (extincteurs, électrique, gaz) font l’objet de contrôle régulier.
Des consignes précises relatives à l’hygiène et à la sécurité du personnel sont définies afin de prévenir tout risque pouvant nuire à la sécurité des personnes et à la qualité des analyses. La prise en charge des patients respecte les mesures de sécurité et de confidentialité.
Les moyens mis en œuvre sont multiples :
-
Affichage des consignes de sécurité
-
Liste et conditions de stockage des produits dangereux
-
Règles de tri et élimination des déchets
-
Lavage des mains et règles d’hygiène et dispositifs appropriés
-
Contrôles bactériologiques des surfaces
-
Salle de détente pour le personnel en dehors des secteurs techniques
-
Vestiaires du personnel avec casier individuel sécurisé dont l’emplacement respecte le principe de la marche en avant (se changer avant d’accéder aux secteurs techniques).
-
Sanitaires pour le personnel, distincts des sanitaires « clients »
-
Plan de désinfection des locaux avec prise en compte des zones à risques.
-
Limitation et contrôles des accès aux secteurs techniques
Le personnel est sensibilisé au risque incendie.
Le personnel technique est formé aux premiers soins et/ou soins d’urgence.
Matériel de mesure et d’essais
Le matériel est installé par le fournisseur ou le fabricant qui est responsable de sa mise en service et de son bon fonctionnement. Un dossier de vérification de méthode (portée flexible A) est constitué par le laboratoire et le personnel technique concerné, avec la preuve des essais de qualification à la mise en service.
La maintenance préventive des équipements est réalisée par le fournisseurs suite à des visites de contrôle contractuelles sont réalisées par le fabricant ou son mandataire qui qualifie le matériel après intervention afin de maintenir le parc matériel en parfait état de fonctionnement. Toutes les actions de maintenance sont enregistrées sur la fiche de vie matériel. Les équipements sont référencés et identifiés.
Le laboratoire a mis en place une surveillance de ses équipements de mesure soumis à métrologie pour leur vérification et étalonnage. Sous la responsabilité du responsable qualité, le technicien référent métrologie est responsable de la gestion des équipements soumis à la métrologie. Elle tient à jour la liste des matériels du laboratoire faisant l’objet d’étalonnage et de vérification et en coordination avec l’équipe des correspondants métrologie.
Les grandeurs physiques sont documentées et font l’objet de surveillance étalonnage et/ou vérification régulière : la température, les volumes, la vitesse (accélération) et le temps (centrifugation). Les opérations de vérifications et de raccordements aux étalons de références sont effectuées par des prestataires compétents, chaque fois que possible. Les certificats de vérification et d’étalonnage sont conservés et disponibles.
Processus – LOCAUX ENVIRONNEMENTS
Processus – GESTION DES MATERIELS
Procédures associées :
-
-
Services externes et approvisionnements (Chapitre 4.6 – ISO 15189)
-
Maîtrise des processus achats et approvisionnements
-
-
Le processus de maitrise des achats comprend l’achat de matériel, de consommables, de réactifs et de prestations de services. Les responsabilités en matière d’achats relèvent de la direction du laboratoire. Les points majeurs relatifs à la maîtrise des achats et approvisionnements :
-
identification des fournisseurs potentiels,
-
évaluation et sélection des fournisseurs :
-
élaboration du questionnaire,
-
choix du fournisseur, liste des fournisseurs approuvés
-
traitement des livraisons non conformes
-
suivi et évaluation des fournisseurs (critiques)
-
contrôle des commandes de produits et matériels,
-
enregistrement des informations et suivi des fournisseurs en matière de Qualité (respect des délais, conformité des livraisons, documents techniques à fournir avec les produits,…).
Un suivi des performances des fournisseurs sélectionnés est réalisé par le pilote du processus. La synthèse globale sur l’évaluation des Fournisseurs « critiques » est présentée en revue de direction.
-
-
-
Vérification du produit acheté
-
-
Toute livraison au laboratoire fait l’objet d’un contrôle à réception du produit livré, par rapport au bon de livraison et à la commande initiale.
Toute non-conformité fait l’objet d’un enregistrement et un traitement est effectué par le secteur ayant passé la commande auprès du fournisseur.
La défaillance des fournisseurs est surveillée. Les données d’évaluation, les non conformités sont présentées en revue de direction.
Processus 07 – ACHATS APPROVISIONNEMENTS
Procédure associée :
-
-
Informatique : gestion des informations du laboratoire (chap. 5.10 – ISO 15189)
-
Le système informatique du laboratoire est sous la responsabilité du biologiste. Un contrat de maintenance (télémaintenance et intervention sur site) est établi avec le fournisseur du SIL.
L’accès au système et aux applications est limité par attribution d’un mot de passe et d’un code utilisateur propre à chaque utilisateur. Le système informatique du laboratoire (SIL) est protégé, dans son accès, par des codes utilisateurs et mots de passes individuels. Le biologiste attribue les autorisations d’utilisation du SIL au personnel, selon leur fonction et responsabilités respectives.
Tous les fichiers système, programme et application sont sauvegardés quotidiennement avec les données relatives aux patients sur des supports distincts. Les supports sont stockés dans un lieu permettant de préserver l’intégrité des données à tout moment et la confidentialité. Un registre des pannes, des modifications des paramétrages et des mises à jour des versions est régulièrement tenu.
Processus 13 – GESTION DU SYSTEME D’INFORMATION – SIL
Procédures associées :
8. REALISATION DES ANALYSES – (Chap. 4.4 – 5.4 – 5.5 – 5.6 – 5.7 – 5.8 – 5.9 – ISO 15189)
-
-
Revue de contrat (Chapitre 4.4 – ISO 15189)
-
Détermination des exigences – besoins
-
-
La revue de contrat précise les critères d’acceptation des demandes d’examens. Cette revue fait l’objet d’un enregistrement (SIL).
Dans le cas où un patient se présente sans prescription médicale, le médecin du laboratoire mène une revue de contrats, avant que le laboratoire ne s’engage à réaliser les examens de biologie médicale demandés, ceci afin d’assurer que les exigences implicites et explicites du patient, et les exigences normatives, législatives, réglementaires et les recommandations de bonnes pratiques sont bien prises en compte. Cette revue de contrats a également pour objectif de prouver que le LBM a les ressources et capacités nécessaires, par sa compétence et grâce à l’utilisation de méthodes répondant aux besoins cliniques du patient, pour réaliser les examens de biologie médicale demandés. Cette revue est réalisée lors de la prise en compte de la demande du patient et porte plus particulièrement sur critères suivants : renseignements cliniques pertinents utiles, conditions et délais de réalisation, modalités de communication des résultats, conditions de facturation et accord du patient sur les montants de la prestation de soins. Dans tous les cas le laboratoire incite vivement le patient a consulté son médecin habituel pour une prise en charge médicale efficiente.
-
-
-
Revue des exigences
-
-
Les demandes d’examens (information sur la réalisation des analyses, prélèvement, tarification, délai de rendu des résultats, destinataires, modalités de transmission des comptes rendus d’analyses) sont vérifiées et enregistrées par le secrétariat du laboratoire. Toute modification portant sur la demande d’examen est enregistrée et communiquée au personnel concerné (exemple : examen non réalisé à la demande du patient, …).
En cas de sous-traitance, la secrétaire ou le biologiste informe le patient et s’assure du respect des besoins, attentes et accord de celui-ci.
-
-
-
Communication avec le client
-
-
Le personnel du secrétariat est autorisé à communiquer des informations relatives aux clients, dans le respect des règles de confidentialité. Les commentaires, avis et interprétations des résultats d’analyses sont réservés exclusivement au biologiste. En cas d’incidents ou anomalies (échantillons biologiques non conformes, délai de rendu des résultats non respecté…), le patient et/ou le médecin sont informés, le cas échéant. Une solution est recherchée, des enregistrements appropriés sont effectués.
-
-
Processus pré analytique (chapitre 5.4)
-
Débute à partir de l’interprétation de la prescription médicale nécessaire à la réalisation du prélèvement. La revue de contrat ou revue des exigences est effectuée au cours de cette étape par la secrétaire du laboratoire au moment de l’accueil du patient. Les prescriptions médicales sont accompagnées d’une fiche de suivi médicale permettant l’identification du patient et le recueil d’informations utiles à la réalisation des analyses et la transmission des comptes rendus d’analyses (renseignements cliniques pertinents).
La réalisation du prélèvement est effectuée par des professionnels de santé habilités selon des instructions spécifiques relatives au prélèvement et à la manipulation des échantillons primaires. (ou par le patient : urines, selles, sperme, …). La vérification de l’identité des patients, en cohérence avec les procédures du laboratoire pour prévenir tout risque d’erreur est systématiquement réalisée par le personnel habilité du laboratoire sensibilisé sur ce risque critique (Identitovigilance).
Les fonctions, qualifications et habilitations pour la réalisation des prélèvements sont définies.
Toute demande d’examens est enregistrée sur le système informatique du laboratoire dans un dossier patient informatisé et identifié par un numéro unique. Les documents fournis par le patient sont photocopiés (chaque fois que possible) et joints à la demande d’examens gérée dans le SIL. Tous les documents produits dans le processus de réalisation des analyses ainsi que les échantillons biologiques sont identifiés, étiquetés par un identifiant unique (numérique et code à barres). La communication avec les clients est formalisée et enregistrée à partir d’une fiche de suivi qui accompagne chaque prélèvement. La traçabilité des évènements clés est organisée.
Les documents et produits fournis par le client sont pris en charge dans des conditions optimum de sécurité et d’intégrité. Les documents fournis par chaque patient sont regroupés dans une pochette plastique individuelle et/ou scanner dans le dossier de chaque patient. Les exigences particulières (urgent) sont signalées par un enregistrement. Tout dossier présentant une difficulté détectée après la prise en charge (information incomplète, anomalies d’identification, erreurs, …) fait l’objet d’un traitement particulier.
Processus 03 – PREANALYTIQUE
Procédures associées :
-
-
Processus analytique (chapitres 5.5 et 5.6)
-
Les opérations relatives à la mise en œuvre des techniques analytiques sont réalisées selon les modes opératoires en vigueur au laboratoire et à disposition immédiate du personnel. Les méthodes d’analyses sont évaluées et validées pour les sous-familles d’examens des portées d’accréditation (sous-famille d’examens). Ce principe se développe avec le déploiement progressif de l’accréditation à toutes les activités. La réalisation des analyses se fait par le personnel qualifié et habilité : technicien(ne)s, biologistes.
La performance analytique est contrôlée par un programme de contrôles internes de qualité et par la participation à des programmes d’évaluation externe de la qualité (par comparaisons inter laboratoires). Le laboratoire, chaque fois que cela est pertinent, est en mesure de déterminer l’incertitude de mesure associée aux résultats.
Le contrôle de qualité analytique (interne et externe) avant analyse participe à la validation de la production des analyses, étape sous la responsabilité des technicien(ne)s habilité(e)s et de biologiste. Les matériels utilisés pour la réalisation des analyses sont contrôlés et maintenus selon les préconisations des fabricants.
La qualification du personnel technique est définie.
Les habilitations sont documentées et enregistrées. Les échantillons biologiques sont stockés et conservés après analyse dans des conditions garantissant leur sécurité et leur intégrité, pour permettre d’éventuels examens complémentaires ou vérifier l’identité portée sur les échantillons (Identitovigilance). Notamment pour permettre la réalisation d’analyses complémentaires ou la répétition des analyses. Toute la documentation technique d’origine externe est validée pour application au laboratoire par la direction du laboratoire.
La vérification des équipements de mesure est effectuée périodiquement pour établir la conformité métrologique.
Processus – – ANALYTIQUE
Procédures associées :
-
-
Processus post analytique – Compte rendu de résultats (chapitre –5.7 – 5.8 – 5.9 – ISO 15189)
-
Le biologiste est responsable de la revue (validation biologique) et la mise en forme des comptes rendus de résultats. Il vérifie la conformité des résultats d’analyses par validation biologique informatique et engage sa responsabilité en signant le compte rendu d’analyses. Le biologiste s’assure, également, que les résultats sont lisibles et ne comportent pas d’erreur de transcription. La transmission des résultats d’analyses est effectuée par tous les moyens disponibles de traitement de l’information, à savoir : le courrier électronique, par fax ou par messagerie.
Les comptes rendus de résultats d’analyses sont édités sur papier à entête comportant les mentions réglementaires concernant le laboratoire et le patient. Figurent notamment :
- le libellé des analyses -les résultats d’analyses
- les valeurs de références tenant compte du sexe et de l’âge (chaque fois que cela est pertinent et possible).
- les commentaires éventuels : avis et interprétation, le cas échéant.
- la méthode ou technique utilisée
- le laboratoire sous-traitant éventuel
- résultat partiel ou définitif
Des conventions de preuves sont mises en place avec les médecins destinataires des résultats par transmission électronique. En pratique, tous les résultats d’examens courants (ou de routine) sont communiqués dans la demi-journée suivant le prélèvement de l’échantillon (exception : les prélèvements réalisés en fin de journée et dont la conservation est possible sont traités à j+1, dans la demi-journée suivante).
Processus – – POSTANALYTIQUE
Procédures associées :
-
-
Analyses transmises à des laboratoires sous-traitants (Chapitre 4.5 – ISO 15189)
-
Le Laboratoire peut être amené à transmettre des analyses ne faisant pas partie du champ de l’accréditation ou qu’il ne réalise pas dans les situations suivantes :
-
Systématique :
-
Examens de biologie médicale spécialisés non réalisés au Laboratoire,
-
Optimisation des plateaux techniques avec mise en place de contrat de coopération.
Le patient est informé de la sous-traitance, le cas échéant. Le laboratoire sous-traitant est accrédité cofrac.
La sous-traitance en cas de panne fait l’objet d’un enregistrement dans le dossier du patient.
Processus– ACHATS APPROVISIONNEMENTS
Processus –- POSTANALYTIQUE
Procédure associée :
9. MESURES, ANALYSES ET AMELIORATION (Chapitres 4.8 – 4.9 – 4.10/11/12 – 4.14 – 4.15)
-
-
Généralités
-
La direction du laboratoire inscrit la démarche d’amélioration continue comme une composante majeure de son système de management de la qualité et de sa politique qualité. Les enregistrements systématiques des non conformités, des réclamations, de toute nature, impliquent l’ensemble de l’équipe du laboratoire.
Afin de maîtriser et améliorer les prestations auprès des clients utilisateurs des prestations du laboratoire (patients, médecins, pharmacies, …), une dynamique de progrès, adaptée aux besoins des clients et impliquant le personnel du laboratoire, est mise en place.
Les contrôles et la surveillance de la qualité des processus et des prestations du laboratoire sont établis par le responsable qualité et validés par les biologistes co-responsables.
L’ensemble de l’équipe du laboratoire participe à la dynamique d’amélioration continue qui porte sur les axes majeurs suivants :
-
Amélioration continue des prestations de services associées au compte rendu de résultats :
-
Optimiser les délais de réalisation
-
Assurer des prestations de service performantes et adaptées
-
Avis et interprétation associés aux résultats (chaque fois que possible)
-
Amélioration continue des processus :
-
Diminuer du nombre de non conformités détectées à tous les niveaux de la réalisation des analyses et au niveau des processus supports
-
Analyser les risques et conduire des analyses de tendance
-
Engager des actions correctives et préventives
-
Amélioration continue de la satisfaction des clients :
-
Réaliser régulièrement des enquêtes de satisfaction
-
Favoriser tout retour « client » et l’enregistrement des réclamations
-
Rencontrer les professionnels de santé : infirmières, médecins, pharmaciens, directeurs d’établissements de santé.
-
-
Surveillance et mesure (chapitre 4.8 – 4.9 – 4.14 – 4.15)
-
Satisfaction du client
-
-
Conformément à la politique qualité du laboratoire, les besoins des clients sont recherchés et identifiés grâce à des enquêtes de satisfaction. Les réclamations sont enregistrées et régulièrement exploitées. Toutes ces données sont traitées et analysées au cours des réunions qualité et revues de direction.
Le laboratoire réalise des enquêtes de satisfaction de manière périodique afin de vérifier l’adéquation entre les attentes des clients et les prestations fournies. Les recueils de données sont effectués sous forme :
-
de questionnaire d’évaluation diffusé aux patients, aux médecins et directeurs de cliniques.
-
d’enregistrement des réclamations ou tout retour d’information de la part des médecins, patients, …
Une exploitation et une analyse sont réalisées par le RQ. Les résultats sont diffusés à l’ensemble du personnel pour une meilleure prise en compte des besoins des clients. Les actions correctives et préventives qui en découlent sont mises en œuvre et revues.
-
-
-
Evaluation et audits
-
-
Audits internes : Une procédure définit les modalités de planification, réalisation, enregistrement et exploitation des audits internes. La mise en place, la revue et l’amélioration du système de management qualité du laboratoire est assurée par la réalisation régulière d’audits qualité internes. Ceux-ci portent sur l’ensemble du système qualité, les processus, les documents.
Un planning annuel d’audits internes est défini et géré (KALILAB®). Ce planning tient compte de la complexité des processus ou activité.
Les écarts ou non conformités constatés lors d’un audit interne font l’objet d’actions d’amélioration (curatives, correctives ou préventives) et d’une revue annuelle.
Des audits ponctuels peuvent être déclenchés, le cas échéant, par la direction ou le responsable qualité dès que la situation l’exige.
Les audits sont réalisés par le personnel habilité auditeur interne, le responsable qualité ou par un auditeur externe indépendant (compétences justifiées, vérifiées). Les audits internes sont réalisés par un auditeur indépendant de l’activité auditée. Le personnel ne doit pas auditer ses propres activités.
Les rapports et les constats d’audit sont enregistrés, diffusés aux personnes concernées et régulièrement suivis avec un bilan annuel réalisé dans le cadre des réunions de revue de direction. La direction évalue l’efficacité du système et décide des actions correctives nécessaires à engager dans un délai raisonnable et convenu.